
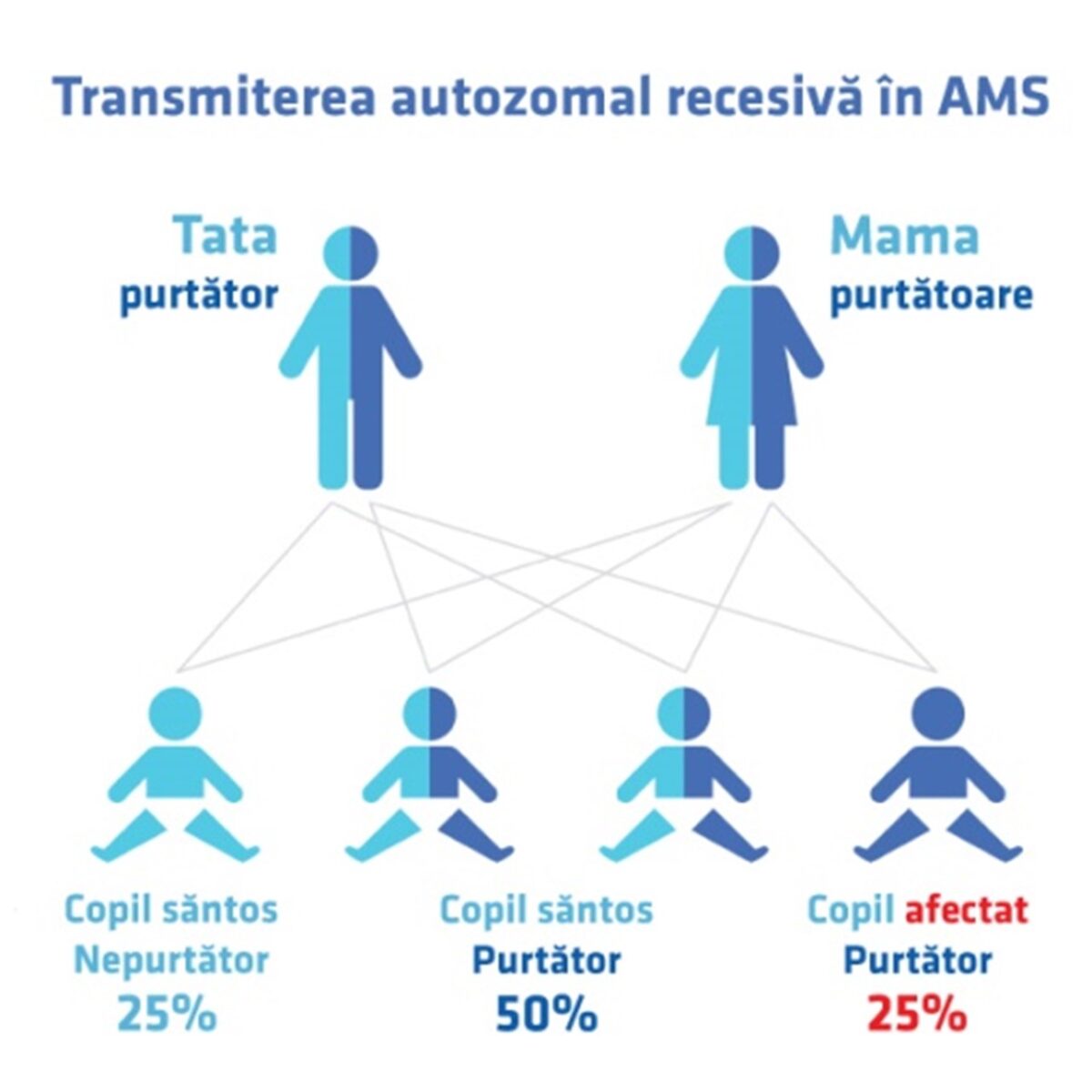
Amitrofia spinală este o afecțiune autozomal recesivă, determinată de mutații la nivelul cromozomului 5 – gena SMN (deletia homozigota a exonului 7). Amiotrofia spinală (AMS) este de mai multe tipuri, în funcție de vârsta debutului simptomatologiei.
AMS se caracterizează în esență prin distrugerea primitivă și moartea prin apoptoza corpilor celulari ai neuronilor periferici din măduva spinării și cei ai nucleilor motori ai trunchiului cerebral.
Tip I | Tip II | Tip III | Tip 0 |
<6l | <18l | >18l | prenatal |
Nu stau in sezut fara sprijin | Nu pot merge fara sprijin | Achizitia mersului | Fara achizitii motorii |
Deces ~2a | Deces dupa 2 ani | Deces in perioada de adult | Deces in primele 3l |

AMS tip I (sau boala Werdnig - Hoffmann)
Debutul este sub 6 luni de viață, având achiziții maxime susținerea capului.
Clinic – scăderea motilității la nivel proximal al membrelor, mai accentuat la cele inferioare.
Sugarii au mișcări active segmentare destul de sărace. Deficitul se generalizează în câteva săptămâni și pierd abilitatea de a-și susține capul sau chiar nu au deloc această achiziție.
Paralizia muschilor intercostali este foarte frecvantă și omniprezentă – apare o deprimare paradoxală a cutiei toracice în inspir, iar abdomenul se expansionează – respirație paradoxală.
Masticația și suptul sunt deficitare – obosesc repede, ulterior refuză alimentarea și scad în greutate.
Reflexele osteo-tendinoase (ROT) sunt abolite sau chiar inexistente.
Fasciculațiile la nivelul limbii sunt prezente in majoritatea cazurilor.
Cognitiv, bebelușii prezintă achiziții psihice în pofida faptului că nu au achiziții motorii.
AMS tip II (forma intermediară)
Debutul este intre 6 si 18 luni, iar achiziția maxima motorie este poziția șezând fără sprijin.
Clinic – Tonus muscular scăzut încă de la naștere sau în primele luni de viață, aceștia pot prezenta discrete și lente achiziții motorii apărând astfel întârzierea în achizițiile motorii. La fel ca si in AMS tip lI este prezentă hipotonia, dar deficitul motor ușor mai puțin intens – reușește să achiziționeze șezutul, de obicei cu sprijin în trepied, pot duce obiecte la gură, dar rar pot ridica brațele deasupra capului.
Reflexele osteo-tendinoase sunt întotdeauna abolite, cele de la nivelul membrelor superior pot fi regăsite dar se pierd ulterior.
Tulburări de deglutiție nu sunt frecvente, dar apare deficit al mușchilor masticatori.
Fasciculațiile limbii sunt prezente in aproximativ 70% din cazurile cu AMS tip II.
AMS tip III (sau boala Kugelberg-Welander)
Debutul este după vârsta de 18 luni – poate chiar doar la vârsta adultă, cu achiziții motorii maxime având chiar și mersul independent.
Clinic – afectare pulmonara cu impact scăzut pentru calitatea vieții fata de tipurile I si II. Scolioza este problema majora la jumătate din acești pacienți, nu este atat de severa ca la cei cu AMS tip II. Subluxația de sold, în schimb, este frecventă.
AMS este o boala ce necesită echipă multidisciplinară pentru o calitate a vieții cât mai bună. Sunt implicați medici de neurologie pediatrica, pediatrie, recuperare medicală si kinetoterapie, ortopedie pediatrică, genetică – sfat genetic pentru părinți, psihoterapie de asemenea este necesară.
Evaluarea neurologică este foarte importantă pentru o diagnosticare cât mai timpurie. Cu cat mai repede cu atat mai multe beneficii asupra calității vieții. Medicul eliberează un voucher pentru testare genetica gratuită, în baza simptomatologiei în urma consultului, iar după confirmarea acestuia urmează pașii pentru inițierea tratamentului. Este foarte important faptul ca, aceasta boala, deși este rara, are tratament .
Risdiplam (Evrysdi, Roche și PTC Therapeutics) este prima terapie cu administrare orală pentru AMS, indicată în cazul adulților și copiilor cu vârsta mai mare de 2 luni, aprobată de Food and Drug Administration (FDA). Risdiplam reprezintă una dintre puținele opțiuni terapeutice pentru atrofia musculară spinală. În prezent, moleculele cu indicație în AMS sunt Zolgensma (onasemnogene abeparvovec-xioi), prima terapie genică indicată în atrofia musculară spinală pentru cei cu vârste mai mici de 2 ani și Spinraza (nusinersen), prima terapie modificatoare de boală aprobată în 2016 de FDA pentru tipurile I-III de AMS.
Eficiența tratamentului cu Risdiplam a fost efectuata cu ajutorul a 4 studii
clinice, FIREFISH, SUNFISH, JEWELFISH și RAINBOWFISH. Aprobarea medicamentului a survenit în urma rezultatalor studiilor FIREFISH și SUNFISH, care au evaluat evoluția pacienților diagnosticați cu AMS de tip I (forma severă infantilă, boala Werdnig-Hoffmann) și respectiv, AMS de tip II (forma intermediară, infantilă cronică) sau AMS de tip III (forma uşoară, juvenilă, boala Kugelberg-Welander).
Studiul FIREFISH s-a realizat pe 21 de participanți cu vârste cuprinse între 1 și 7 luni, diagnosticați cu atrofie musculară spinală tip I și la care au fost identificate 2 copii ale genei SMN2. Risdiplam a fost administrat zilnic, timp de 4 săptămâni, în doze diferite. Datele obținute au relevat următoarele aspecte:
- 41% dintre pacienți au reușit să stea in șezut mai mult de 5 secunde fără suport la 12 luni de tratament;
- 81% dintre pacienți au supraviețuit după 23 de luni de tratament fără a necesita ventilație mecanică;
- proteina SMN a înregistrat o creștere de 6,5 ori la pacienții care au primit tratament cu Risdiplam, față de nivelul de bază.
Beneficiile tratamentului cu Risdiplam pentru AMS de tip II și III au fost evaluate concomitent în cadrul studiului dublu-orb SUNFISH, desfășurat în două etape. În prima parte s-a stabilit doza optimă de administrare, urmând ca în etapa II să fie analizată eficiența tratamentului cu Risdiplam. Cea de-a doua parte a studiului a inclus 180 de pacienți între 2 și 25 de ani, cu diagnostic de AMS tip II sau III. Obiectivul principal a constat în măsurarea funcției motorii cu ajutorul scalei Motor Function Measure 32 (MFM-32) după 1 an de tratament cu Risdiplam, comparativ cu administrarea de placebo. Rezultatele au fost după cum urmează:
- Cei care au primit Risdiplam au înregistrat o creștere de 1.36 ori mai mare a funcției motorii comparativ cu grupul placebo, unde pacienții au avut funcția motorie redusă față de punctul de plecare;
- Cel mai bun răspuns la tratament conform MFM-32 a fost observat în grupul de pacienți tineri (2-5 ani);
- 57,1% dintre pacienții în grupa de vârstă 18-25 de ani au stagnat sub tratamentul cu Risdiplam, fără a prezenta regres dpdv al deficitului motor, comparativ cu doar 37,5% din grupul de placebo.
În prezent, atrofia musculară spinală beneficiază de 3 opțiuni terapeutice (toate acestea având aprobare FDA).
- În 2016 în S.U.A. s-a implementat Spinraza (Nusinersen), prima terapie modificatoare de boală indicată în AMS tipurile I-III. La fel ca și Risdiplam, Nusinersen acționează prin reglarea expresiei ARN-ului mesager pentru pentru gena SMN2. Diferența majoră constă în modul de administrare. Tratamentul cu Spinraza presupune injecții administrate intratecal (în lichidul cefalorahidian care înconjoară măduva spinării) pentru tot restul vieții pacienților;
- Zolgensma a fost aprobată de FDA în 2019 și este indicată la pacienții cu vârste sub 2 ani cu atrofie musculară spinală, având mutație bialelică la nivelul genei SMN1. Zolgensma este administrat o singură dată, intravenos și acționează prin introducerea genei SMN1 funcționale în corp. Astfel, proteina SMN va fi sintetizată corespunzător;
- Risdiplam, singura terapie cu administrare orală, indicată în tipurile I-III de AMS, aprobată de FDA în 2020.
Zolgensma este a 2-a terapie genică aprobată de FDA, după Luxturna.
Din fericire, astazi exista si in tara noastra tratamentul ideal pentru acest tip de boala.
Exista asociații care sprijină părinții pentru a putea gestiona cât mai bine nevoile celui mic.
AMS afectează aproximativ 1 din 10.000 nașteri vii la nivel mondial, fiind cea mai întâlnită cauză genetică de deces în rândul sugarilor. Boala este cauzată de o mutație/deleţie la nivelul genei SMN1, ceea ce conduce la niveluri scăzute ale proteinei de supraviețuire a neuronilor motori (SMN). Această proteină se regăsește în întreg organismul şi are un rol vital în funcționarea sistemului nervos. Tratamentul este vital pentru evoluție cat mai buna!

Dr. Mădălina Lascu
Medic specialist neurologie pediatrică
Bibliografie
- Neurologie Pediatrica – Conf.Dr.dana Craiu, Catrinel Iliescu Editura Universitara “Carol davila” București 2013
- https://raportuldegarda.ro/atrofia-musculara-spinala-diagnostic-tratament-precoce/
- https://assets.roche.com/f/172042/x/be135a3d46/pr_220608_roche_medicament-atrofie-musculara-spinala.pdf
- http://www.saptamanamedicala.ro/articole/Terapie-inovatoare-disponibila-pentru-pacientii-cu-atrofie-musculara-spinala-din-Romania









